-

-
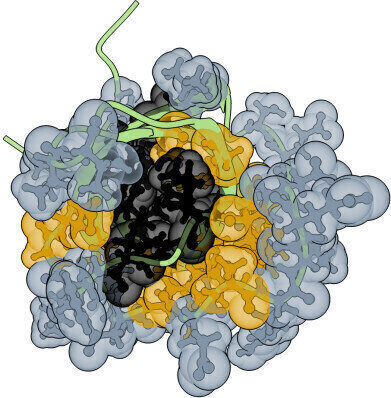 The core (gold and black) and surface (silver) residues that were randomised. Credit: Albert Escobedo/Centro de Regulación Genómica
The core (gold and black) and surface (silver) residues that were randomised. Credit: Albert Escobedo/Centro de Regulación Genómica
Research news
One billion-year-old rules of protein stability revealed as ‘like Lego not Jenga’
Jul 25 2025
Huge experiment uncovers rules governing protein stability, opening up novel opportunities for drug and enzyme design
A vast experimental study has revealed underlying rules that govern protein stability, offering a potentially transformative approach to the design of drugs, enzymes and other biologically active molecules.
Proteins – the molecular machinery of life – are constructed from 20 types of amino acid. Even a modestly sized protein of 60 amino acids could, in theory, be assembled in 10⁷⁸ different combinations – a quinquavigintillion – which is a number similar to the total number of atoms estimated to exist in the entire observable universe. This combinatorial explosion has long raised a fundamental question: how has evolution navigated such an immense search space to identify the rare combinations that produce stable, functional proteins?
The conventional wisdom about proteins, long held by biologists, has been that they behave like the structural foundation of a building – so densely packed that a single modification could lead to unpredictable shifts and render the entire molecule unstable. According to this view, most mutations would result in a collapse of the folded structure, but this would make the evolutionary process that produces viable novel proteins seem implausibly inefficient.
However, researchers at the Centre for Genomic Regulation (CRG) in Barcelona, Spain and the Wellcome Sanger Institute in Hinxton, near Cambridge in the UK, have reported findings that contradict this traditional view. They systematically mutated a human protein domain known as FYN-SH3 – a small, evolutionarily conserved region involved in cell signalling – generating and testing hundreds of thousands of variants to assess their ability to fold and retain function.
The results indicate that SH3 domains retain their integrity across a surprisingly wide range of amino acid substitutions. Although certain key amino acids in the core are essential, most alterations – even in buried regions – do not compromise protein stability. This suggests that the evolutionary path to functional proteins may not be as precarious as previously thought.
“Our data challenges the dogma of proteins being a delicate house of cards.
“The physical rules governing their stability is more like Lego than Jenga, where a change to one brick threatening to bring the entire structure down is a rare, and crucially, predictable phenomenon,” said Dr Albert Escobedo, first author and postdoctoral researcher at CRG.
The team used the resulting dataset to train a machine-learning algorithm which was capable of predicting the stability of other SH3 domains. They then tested the model against more than 51,000 naturally occurring SH3 sequences drawn from across the natural world, including bacteria, plants, insects and vertebrates. Remarkably, the model correctly identified almost all of these domains as stable – even when the sequences diverged significantly from the human version.
SH3 domains are thought to have diversified over the past billion years, since the emergence of early multicellular life. The research indicates that shared biochemical rules have underpinned protein evolution throughout this entire timespan.
“Evolution didn’t have to sift through an entire universe of sequences. Instead, the biochemical laws of folding create a vast, forgiving landscape for natural selection,” says Dr Escobedo.
Protein engineering, by contrast, has until now proceeded with extreme caution. Companies typically alter only a few amino acids at a time and screen thousands of variants to identify rare improvements. This iterative approach is slow and costly, particularly when designing novel enzymes or therapeutic proteins.
The confirmation that protein stability depends on a limited set of rules may allow for more ambitious strategies. For example, therapeutic enzymes often provoke immune reactions due to their surface features. Efforts to re-engineer these surfaces have been constrained by the risk of destabilising the overall structure. With greater predictive power, researchers can now design proteins in silico that might have multiple simultaneous modifications and select only the most promising candidates for laboratory validation.
“The ability to predict and model protein evolution opens the door to designing biology at industrial speed, challenging the conservative pacing of protein engineering,” said Professor Ben Lehner, a research professor at CRG and corresponding author of the study, who also holds an affiliation with the UK’s Wellcome Sanger Institute.
The findings are likely to accelerate the development of enzymes for sustainable industrial processes, as well as next-generation biologics with improved therapeutic performance and safety.
For further reading please visit: 10.1126/science.adq3948
Digital Edition
Lab Asia Dec 2025
December 2025
Chromatography Articles- Cutting-edge sample preparation tools help laboratories to stay ahead of the curveMass Spectrometry & Spectroscopy Articles- Unlocking the complexity of metabolomics: Pushi...
View all digital editions










Events
Jan 21 2026 Tokyo, Japan
Jan 28 2026 Tokyo, Japan
Jan 29 2026 New Delhi, India
Feb 07 2026 Boston, MA, USA
Asia Pharma Expo/Asia Lab Expo
Feb 12 2026 Dhaka, Bangladesh




